|
||||||||
| HOME | METHODS | LINKS | NEWS | TIPS | SOFTWARE | FORUM | CONTACT | SEARCH |
| DNA methylation analyis using restriction enzyme digestion |
|
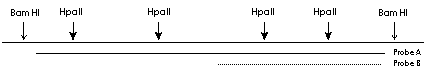
his is a classical method of methylation analysis based on the property of some restriction enzymes to be unable to cut methylated DNA. Since in eukaryotic DNA (or actually in mammalian DNA) only cytosine in CG context can be methylated, the restriction enzymes with CG sequences within their restriction sites come in question. Two classical enzyme pairs are HpaII - MspI (CCGG) and SmaI - XmaI (CCCGGG). Since the second recognition sequence is much more rare than the first one I will concentrate on the HpaII-MspI pair. Both enzymes recognize CCGG sequence, however HpaII is unable to cut DNA when the internal cytosine is methylated. This property makes HpaII-MspI pair to a valuable tool for rapid methylation analysis. This method has some weak points. First is that not all CG are located within CCGG sequences, that means many potential methylation sites will be overlooked. Another problem is a necessity of a Southern blot hybridisation which is not uncomplicated. However it is still a good method to get a first idea if the methylation is involved in the phenomenon you observe. Before you proceed with a methylation analysis you have to analyse your sequence very carefully. If you'll just take genomic DNA, digest it with HpaII, blot and hybridise with selected probe the result will be very complicated. With 200 bp probe you can obtain myriad of bands which you will not be able to interpret. Therefore first of all define the region of interest flanked with restriction sites for CG methylation insensitive enzymes (BamHI for example), and containing not more than 5-6 sites for HpaII. The probe used for Southern blot hybridisation should be located within this region and cover it completely or partially (see the figure).
Even in this case when you will use the probe A the number of fragments will be relatively high (try to count them if you make complete digestion with BamHI and then - incomplete with HpaII). Probe B will give you less complex picture, but actually the same information. Another problem is the size of the fragments you usually obtain. If you have a CG rich region, then HpaII restriction fragments will be in the range 100-500 bp. For this fragment range you will need 1.2% agarose and such gels are relatively difficult to blot. However this method seems to be simpler and faster to establish than bisulphite treatment. Protocol
Do not expect to obtain a clear cut results. Usually cell lines and especially tissues are polyclonal and contain mixed methylation patterns, therefore your band pattern will be also complex.
|
| \METHODS\DNA methylation analysis\Methylation analyis using restriction enzyme digestion |
| © Copyright 1999-2006 Alexei Gratchev. All rights reserved. |